제품특징
The only user-friendly
phylogenetic analysis software
Align sequences, build and view phylogenetic trees using your choice of algorithm,and edit the images for publishing - all in one program.
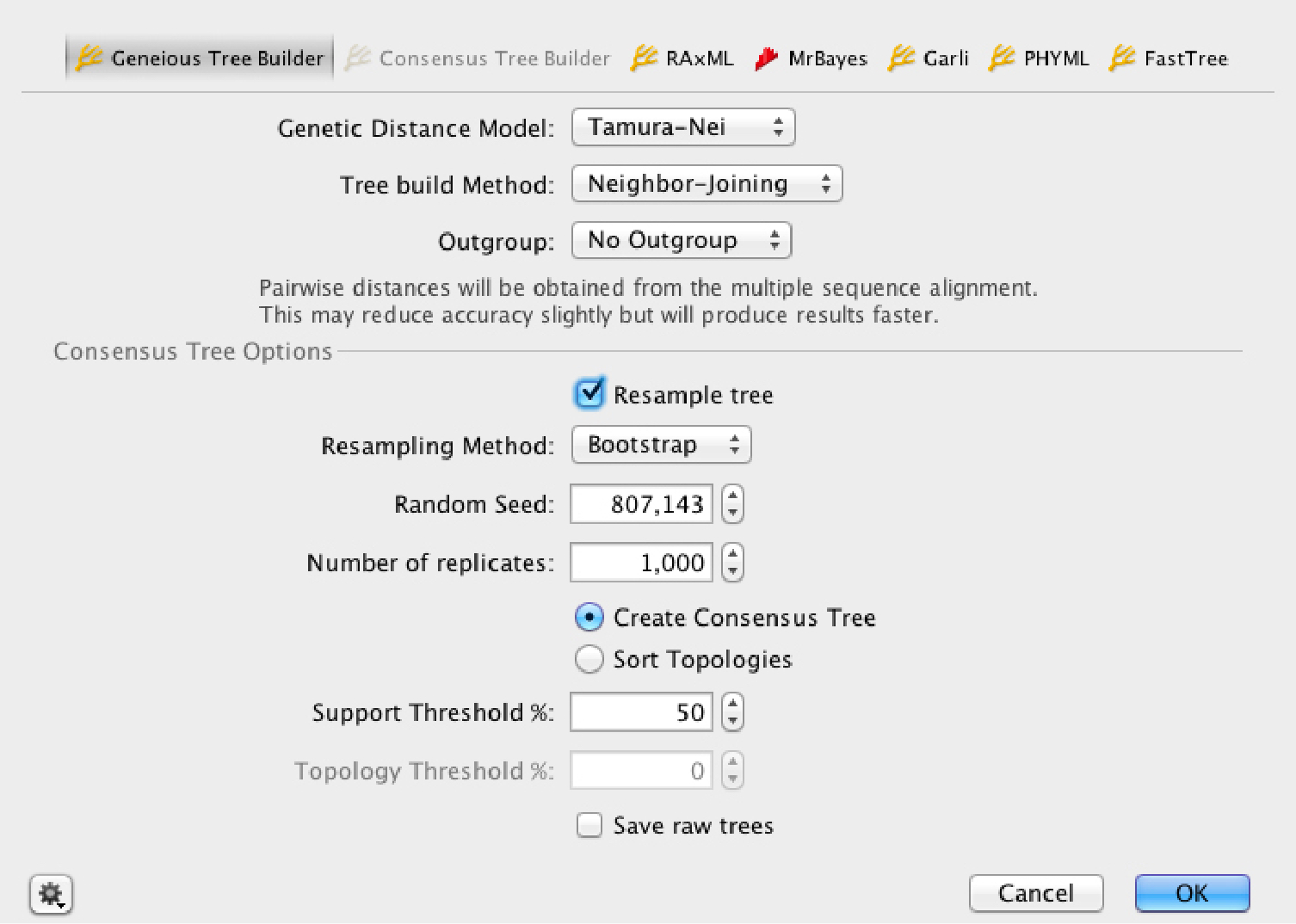
Built-in likelihood, distance and
bayesian tree building methods
Interactive distance matrix viewer는 Phylogenetic 분석을 위해 빠르고 유의한 통계를 제공합니다.
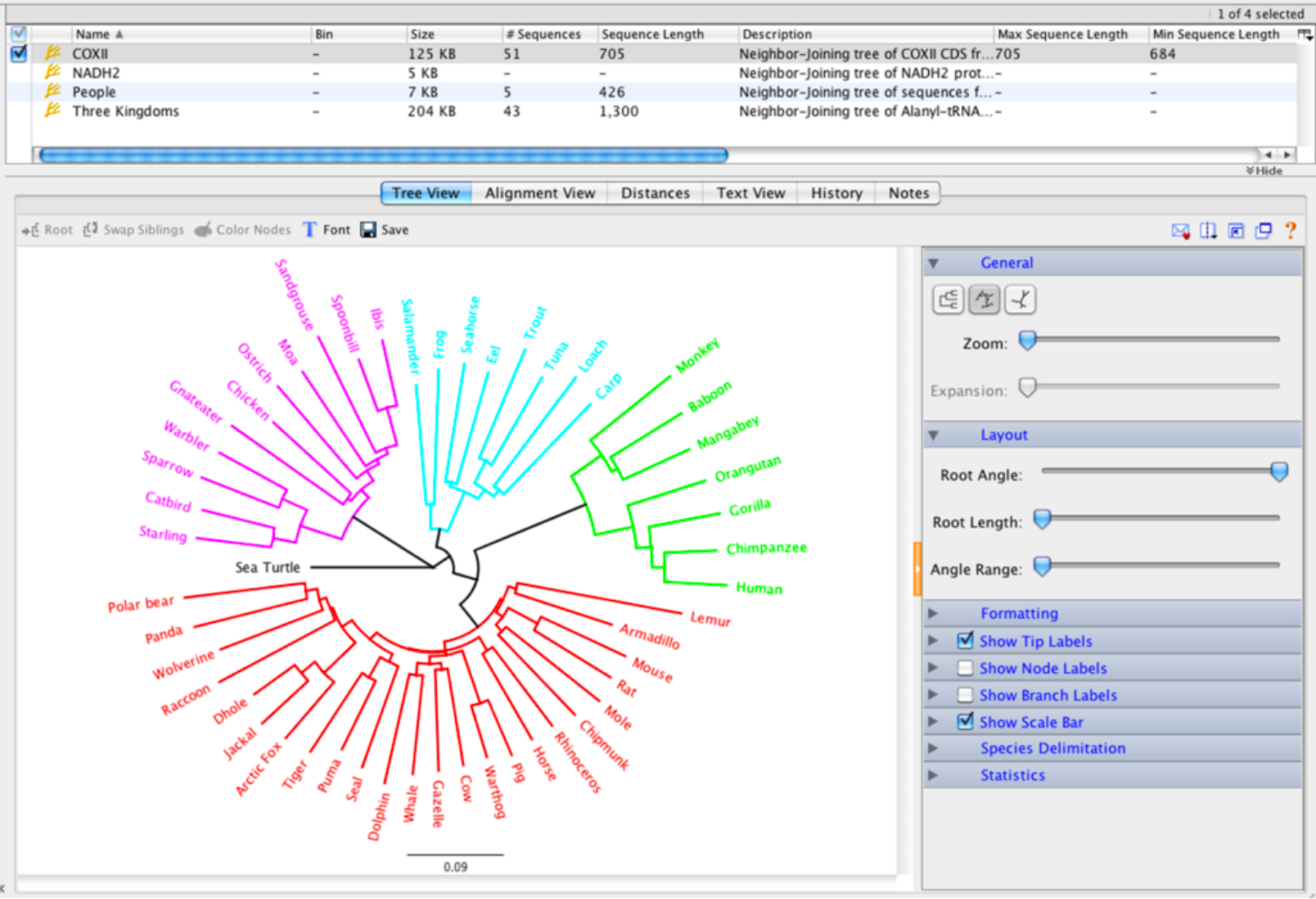
Tree building and viewing
without juggling files
클릭 한번으로 다중서열 정렬 데이터와 알고리즘을 선택하기만 하면 Tree가 생성됩니다.
Your choice of tree building algorithms
- Neighbor Joining – 빠르고 간단한 neighbor-joining methodology를 사용하여 몇 초 만에 많은 수의 taxa를 위한 가이드 트리를 제작
- UPGMA – Phylogenetic reconstruction을 위한 단순하고 빠른 hierarchical clustering method
- MrBayes – Phylogeny의 bayesian estimation을 위해 MrBayes 플러그인을 통하여 로컬 컴퓨터에서 실행
- PAUP* – PAUP* 플러그인을 통하여 GUI 인터페이스로 Maximum Parsimony, Maximum Likelihood를 수행(이 플러그인을 활용하려면 PAUP*을 보유하거나 구매해야함)
- PhyML – PhyML 플러그인을 통해 maximum likelihood를 최대로 빠르게 구현함
- GARLI- Rapid Likelihood Inference를 위한 genetic 알고리즘 제공
- RAxML- Randomized Axelerated Maximum Likelihood 제공
- FastTree – Nucleotide 또는 protein 서열들의 정렬을 통한 approximately-maximum-likelihood phlogenetic tree 제공
Your choice of tree building algorithms
디스플레이 설정을 통하여 branch label, node label, end label, tree shape, tree scale 또는 clade의 필요한대로 정확하게 바꿀 수 있습니다.
Tree를 구성하면 svg, pdf 또는 eps와 같은 고품질 벡터 형식으로 저장하여 게시하거나 인쇄하여 벽에 붙일 수 있습니다.